人工知能を用いた天然物の迅速構造予測
AI-based Rapid Structure Prediction of Natural Products
天然物探索を専門とする研究者にとって,化合物の構造を早い段階で予測するスキルは重要だ。経験を積んだ研究者になると,1H NMRを眺めただけで化合物のおおよその構造を予測できるようになるが,それでも普段扱っている系統と別のタイプの天然物に出会うと予測の正確性は鈍る。
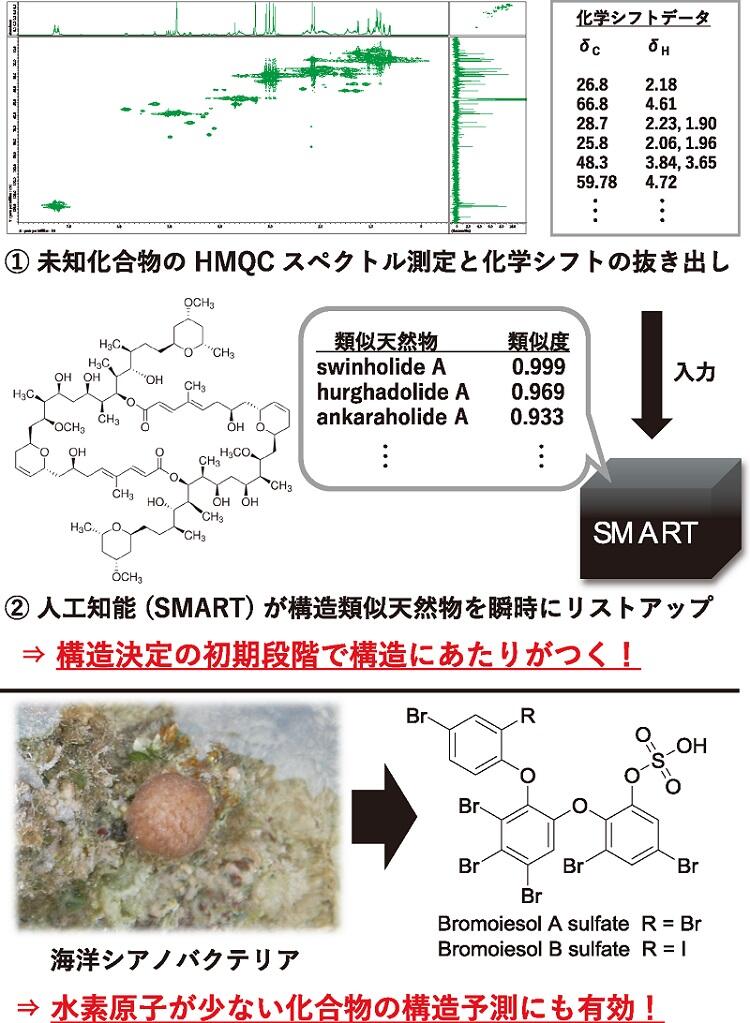
近年,カリフォルニア大学サンディエゴ校のGerwick教授が開発したSmall Molecule Accurate Recognition Technology(SMART)1)は,これまで熟練した研究者の経験と勘に頼ってきたNMRによる構造予測を,人工知能が代替する手法である。使い方はいたって簡単で,Webサイト2)にアクセスし,手持ちの化合物の1H化学シフトと,それに直接結合している13C化学シフトを入力するだけである。結果は瞬時に得られ,構造の類似性の高い天然物がリストアップされる。構造のあたりがつくことで,筆者の所属研究室では,経験の浅い学生でも効率的に構造決定を進めることが可能となった。さらに,水素原子が少なく通常のNMR解析では構造予測が難しいタイプの化合物に対しても,本手法が有効であることを筆者らは見いだしている3)。混合物に対する解析例も報告4)されており,今後の発展・応用に注目が集まる。
1) R. Reher et al., J. Am. Chem. Soc. 2020, 142, 4114.
2) https://smart.ucsd.edu/classic
3) A. Ebihara, A. Iwasaki et al., J. Org. Chem. 2021, 86, 11763.
4) J. Lee et al., ACS Omega 2020, 5, 23989.
岩﨑有紘 慶應義塾大学理工学部化学科